The discovery of a family with extremely Short QT Syndrome interval
Dr. Preben Bjerregaard
Shalon
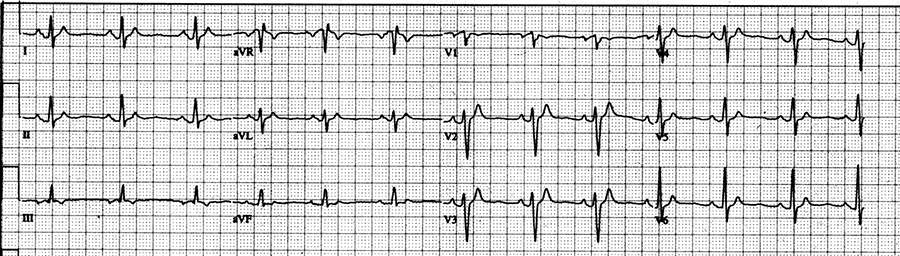
May 5th, 1999 When a 17-year old girl returned to my clinic at Saint Louis University Hospital, Saint Louis MO, USA, and had an ECG recorded following DC cardioversion of new onset atrial fibrillation, I saw for the first time a QT interval in an ECG of a human being of only 250 msec at a heart rate of 75 beats per minute:
QT interval in an ECG of a human being of only 250 msec at a heart rate of 75 beats per minute.
February 16, 1999 The girl initially presented to Anderson Hospital, Maryville, Madison County , Illinois with abdominal pain due to cholelithiasis and underwent laparoscopic cholecystectomy. During the procedure she developed atrial fibrillation with fast ventricular response and heart rates as high as 200 beats per minute accompanied by respiratory distress. Immediately after the procedure she underwent DC cardioversion with restoration of normal sinus rhythm. She had no prior history of atrial fibrillation, palpitations or any other cardiac symptoms.
March 28, 1999 She suddenly felt her heart beating fast, and again an ECG showed atrial fibrillation.
April 14, 1999 While still in atrial fibrillation, she was seen for the first time in my clinic, when the heart rate in her ECG was 120 beats per minute with a QT interval 240 msec, and very suspect for being short.
May 5, 1999 When she returned to my clinic in normal sinus rhythm following her second successful DC cardioversion, the QT interval was still very short at 250 msec, and this time at a heart rate of only 75 beats per minute (QTc: 280 msec) and therefore extremely short.
June 6th, 1999 At her next clinic visit she was accompanied by her mother and older brother, and on the suspicion of a hereditary disorder I asked permission to see their ECG’s, and both of them had very similar short QT intervals.
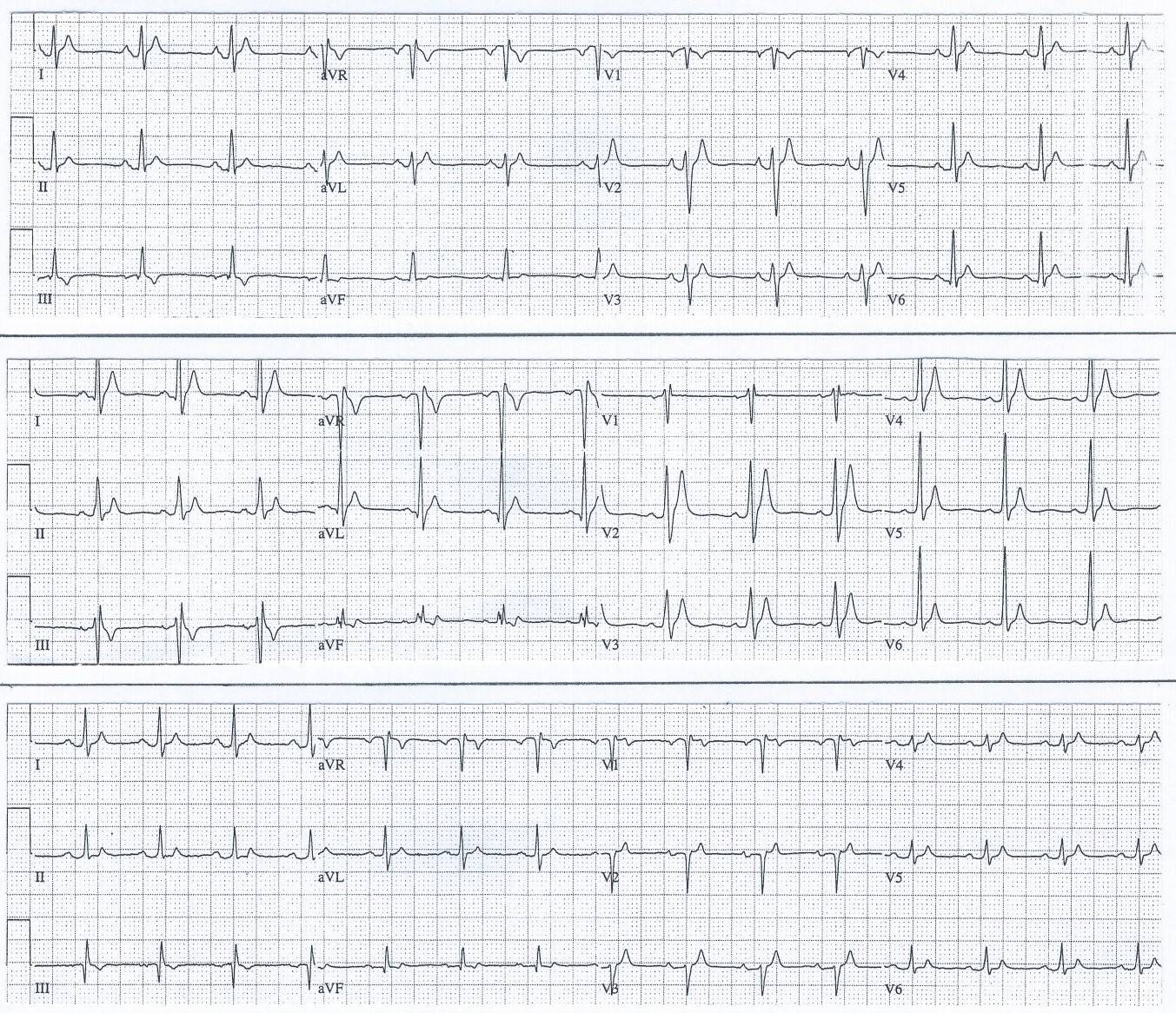
ECGs from first family diagnosed with Short QT Syndrome
ECGs from first family diagnosed with Short QT Syndrome
Family ECG Explained:
TOP TRACING: 12 lead ECG from 17-year-old female with QT-interval 280 ms at HR 76 bpm (QTc 321 ms).
MIDDLE TRAC: 12 lead ECG from her 21-year-old brother with QT-interval 260 ms at HR 79 bpm (QTc 298 ms)
BOTTOM TRACING: 12 lead ECG from their 51-year-old mother with QT-interval 270 ms at HR 88 bpm (QTc 327 ms)
At that moment I knew I had achieved the dream of any scientist – to know something that nobody else has ever known.
(Later it turned out that the patient’s maternal grandfather, who had immigrated from Italy, also had similar very short QT interval, while her father had a normal QT interval. (The girl was later married and got a girl, who also has short QT)).
I discussed my observations with Dr. Ihor Gussak, an experienced basic electrophysiologist, who had just joined our faculty at Saint Louis University Hospital, and he agreed after meticulous search of the medical literature, that we were facing a very unique phenomenon with no prior mentioning. We considered a possible relationship between the short QT and atrial fibrillation, and we were concerned that such a short QT interval could lead to more life-threatening heart rhythm disturbances, possible ventricular fibrillation and sudden cardiac death. We just did not think we had enough for a publication yet.
In a telephone conversation with Dr. Joseph Brugada from Barcelona, Spain, Dr. Gussak mentioned our findings, and to much surprise, Dr. Brugada had an ECG similar to ours. It was from a 38 year old woman, who had been scheduled to see him in 1996 because of fainting spells. Unfortunately she had died suddenly during one of her spells a few days prior to her appointment with him. Also he had not been able to find any explanation in the medical literature for a connection between a Short QT and sudden death, but at least we know had circumstantial evidence for such a connection. With our two stories together, we then decided to publish our findings and suggest a possible new syndrome.
December 27, 1999 We submitted our manuscript to CIRCULATION and already by 01.15.2000 we were informed, that it had been reviewed by the editor and one reviewer and was not found to be acceptable for publication in its present form. The reviewer had raised some important issues, but if these issues could be resolved, they would be willing to consider a revised manuscript. The reviewer found it a potentially very interesting observation, and perhaps we had really identified a unique syndrome, but the reviewer was quite concerned about our ECGs: "They look quite touched up, one wonders about an error of the paper speed". The original ECGs were requested, but even after we had send the original ECGs to the editor, there was still some doubt about the paper speed and the paper was rejected. Our interpretation of the rejection was that neither the editor nor the reviewer believed in such a short QT, which had never been described in a human being before.
Gussak I, Brugada P, Brugada J, Wright RS, Kopecky SL, Chaitman BR, Bjerregaard P. Idiopathic Short QT Interval: A New Clinical Syndrome? Cardiology 2000;94(2):99-102
The list of authors had grown to seven. Ihor Gussak and I embrace a group of colleagues, who had all been helping us. Joseph Brugada from The University of Barcelona, Spain had provided ECG and medical history of one of the patients, and Pedro Brugada from the Cardiovascular Research and Teaching Institute, Aalst, Belgium along with R. Scott Wright and Stephen L. Kopecky from The Mayo Clinic, Rochester, Minnesota, USA had all participated in discussions about the possible relationship between a short QT and serious heart rhythm disturbances. Finally, Bernard R Chaitman was included as Chief of Cardiology at the hospital from where the syndrome originated.
In the article we showed the ECG’s of the initial family (the proband, her brother and her mother) and of the 37 year old Spanish woman, which had all earlier been turned down by the editor of Circulation. We also incorporated an ECG rhythm strip from a 4-y-o African-American girl during an episode of brady-arrhythmia accompanied by deceleration-dependent shortening of the QT interval. We realized that we were unable to assess whether or not we were dealing with a new clinical syndrome or simply with an ECG phenomenon of idiopathic short QT interval. We also stated, that if additional supporting clinical data became available, we believed that – parallel to the “long QT syndrome” – the combination of short QT interval and electrical instability could appropriately be named the “short QT interval Syndrome”.
The following 3 years we were waiting for another family with similar findings to show up somewhere in the world, and especially to find out about the danger of having such a short QT interval. Our family was doing well except for occasional episodes of atrial fibrillation.
First description of a high incidence of SCD in families with Short QT Syndrome
Gaita F, Giustetto C, Bianchi F, Wolpert C, Schrimpf R, Riccardi R, Grossi S, Richiardi E, Borggrefe M. Short QT Syndrome. A Familial Cause of Sudden Death. Circulation 2003;108:965-970
March 2003: The suspicion of a new syndrome which was potentially life-threatening was hightened at the ACC meeting in Chicago, where a paper from Ospedale Mauriziano Umberto I, Torino, Italy describing a family with history of sudden cardiac death and Short QT was persented by Dr. Fiorenzo Gaita. It described 16 members from 5 generations of an Italian family, where there were 6 sudden cardiac death victims including one with short QT. Two additiona members of this family had short QT.
In the paper above from Circulation, August 2003 this Italian family was described in more detailles in connection with the first presentation from The University Hospital Mannheim, University of Heidelberg, Mannheim of 23 members of a German family, where 6 had died suddenly at the ages of 3 months, 6, 39, 39 and 49 years old respectively and one unknown. Four family members had ECGs with short QT interval, including one of the sudden cardiac death victims.
This article also included the first data from electrophysiologic studies of patients with Short QT Syndrome including the very short ERP’s in such patients both in the atria and the ventricles, and also a high incidence of VF (2 out of 4) in these patients during simple catheter positioning in the right ventricle.
Finally the minimal shortening of the QT interval during stress testing was pointed out by the authors, who had implanted ICD’s prophylactically in 3 patients with Short QT Syndrome.
Based upon this excellent and very comprehensive publication by Gaita et al. in Circulation August 2003 of two families with high incidence of sudden cardiac death in the setting of familial short QT interval, there was finally enough clinical data to support our initial findings suggesting a new clinical syndrome: Short QT Syndrome.
The first detailed description of ICD implantation in patients with Short QT Syndrome
Schrimpf R, Wolpert C, Bianchi F, Giustetto C, Gaita F, Bauersfeld U, Borggrefe M. Congenital Short QT Syndrome and Implantable Cardioverter Defibrillator Treatment: Inherent Risk for Inappropriate Shock Delivery. J Cardiovasc Electrophysiol 2003;14:1273-1277
September 29,2003: Press release from Saint Louis University Hospital
This paper describes in more detail the experience of ICD implantation in 5 patients from the Italian and German families mentioned above. Due to T-wave over-sensing, inappropriate shocks were delivered in 3 out of the 5 patients shortly after implantation. Programming lower sensitivities and decay delays prevented further inappropriate discharges.
Incidentally the over-sensing of the T wave lead to the observation, that patients with short QT syndrome often have prominent peaked T waves.
First mutation found in families with Short QT Syndrome
Brugada R, Hong K, Dumaine R, Cordeiro J, Gaita F, Borggrefe M, Menendez TM, Brugada J, Pollevick GD, Wolpert C, Burachnikov E, Matsuo K, Wu YS, Guerchicoff A, Bianchi F, Giustetto C, Schrimpf R, Brugada P, Antzelevich C. Sudden Death Associated With Short-QT Syndrome Linked to Mutations in HERG. Circulation 2004,109:30-35
It was from the Masonic Medical Research Laboratory, Utica, NY the first report about a mutation linked to Short QT Syndrome came in January 2004.
Genetic testing was performed in members of the previously described two families from Italy and Germany in addition to a family from the US consisting of a 51 y.o. father with aborted SCD and his 20 y.o. son, who both had short QT intervals < 300 msec.
In the German family they identified a missense mutation (c to g substitution at nucleotide 1764) in KCNH2 and in the Italian family a different missense mutation in the same residue (c to a substitution at nucleotide 1764). Both mutations, however, substituted the asparagine (N) at codon 588 in KCNH2 protein for a positively charged lysine (K) resulting in the same amino acid change (N588K) in the S5-P loop region of the cardiac IKr channel HERG (KCNH2). No mutation was found in the US family.
The net effect of the mutation is to increase the repolarization currents active during the early phases of the AP, by eliminating current inactivation leading to loss of normal rectification of the current at plateau voltages with abbreviation of the action potential and thus abbreviation of the QT interval.
The N588K missense mutation was also shown to reduce the affinity of the channel for drugs with class III antiarrhythmic action such as Sotalol and Dofetilide. Sotalol did not prolong the QT interval in patients with short QT.
This was the first description of a genetic abnormality responsible for cases of SQTS – later referred to as SQT1.
The first detailed description of pharmacological treatment of patients with Short QT Syndrome
Gaita F, Giustetto C, Bianchi F, Schrimpf R, Haissaguerre M, Calo L, Brugada R, Antzelevitch C, Borggrefe M, Wolpert C. Short QT Syndrome: Pharmacological Treatment. JACC 2004;43:1494-1499
Six members from the previously mentioned German and Italian families were tested with Flecainide, Sotalol, Ibutilide and Hydroquinidine. Flecainide, Sotalol and Ibutilide did not produce any significant QT prolongation. Only hydroquinidine prolonged the QT interval from 263 +/- 12 msec to 362 +/- 25 msec with prolongation of the ventricular effective refractory period to > 200 msec and VF no longer inducible. The slight prolongation of the QT interval following Flecainide was mainly due to QRS prolongation.
The lack of QT prolongation following selective IKr-blocking agents like Ibutilide and Flecainide suggest that the (N588K) mutation in the KCNH2 channel in patients with SQT1 may have caused loss of some of the physiologic regulatory mechanisms, and the ion channel is no longer sensitive to a drug that normally has a specific action on it.
Quinidine was recommended as the drug of choice for medical therapy while Flecainide because of some increase in the effective refractory period could be the second choice.
This study was in a very small group of patient with a single mutation, and the results, therefore, not necessarily applicable to future patients with Short QT Syndrome and other mutations, who might respond to these antiarrhythmic drugs differently.
Li J, McLerie M, Lopatin AN. Transgenic upregulation of IK1 in the mouse heart leads to multiple abnormalities of cardiac excitability. Am J Physiol Heart Circ Physiol 2004;287:H2790-H2802
At the Department of Molecular and Integrative Physiology, University of Michigan Medical School, Ann Arbor, Michigan, the authors produced and characterized the first transgenic mouse model of IK1 up-regulation, which lead to multiple abnormalities in excitability, including pronounced shortening of action potentials and effective refractory period with a short QT interval in the ECG.
The authors felt their data strongly supported the notion that a short QT interval is indeed associated with life-threatening arrhythmias in humans. Thus the transgenic mouse model of IK1 up-regulation might be considered the first experimental model of Short QT Syndrome.
Interestingly enough, in 2005 a group from The University of Pavia, Italy (Silvia G. Priori el al.) found a mutation in the KCNJ2 gene that regulates the Kir2.1 potassium channel (IK1) in a patient with Short QT Syndrome. Since it was the third mutation found in a patient with Short QT Syndrome, it was named SQT3.
First successful treatment of VF by an ICD in a patient with Short QT Syndrome
Schimpf R, Bauersfeld U, Gaita F, Wolpert C. Short QT syndrome: Successful prevention of sudden cardiac death in an adolescent by implantable cardioverter-defibrillator treatment for primary prophylaxis. Heart Rhythm 2005;2:416-417
Case report of a previously asymptomatic 16 y.o. male from the first German family with Short QT Syndrome had received a prophylactic ICD. He had a QT interval of only 248 msec with a QTc of 252 msec. Interestingly, at his initial work-up ventricular tachy-arrhythmias could not be induced during programmed ventricular stimulation. Six month later the ICD saved his life, when he developed ventricular fibrillation during sleep.
The episode occurred during a HR of approximately 60 bpm, initiated by a premature beat with a coupling interval of 180 msec.
First sub-group of Short QT Syndrome presenting as bradycardia in utero.
Hong K, Piper DR, Diaz-Valdecantos A, Brugada J, Oliva A, Burashnikov E, Santos-de-Soto J, Grueso-Montero J, Diaz-Enfante E, Brugada P, Sachse F, Sanguinetti MC, Brugada R. De novo KCNQ1 mutation responsible for atrial fibrillation and Short QT Syndrome in utero. Cardiovasc Res 2005;68:433-440
In Sevilla, Spain a baby girl was born following interruption of the pregnancy at 38 weeks due to severe bradycardia and irregular rhythm, suggesting atrial fibrillation since the 6th month of gestation. At birth the baby was stable at a heart rate of 60 bpm. The ECG showed a short QT interval and atrial fibrillation was confirmed by intracardiac electrography. DC cardioversion was unsuccessful.
By a combined effort at several laboratories headed by the Masonic Medical Research Laboratory in Utica, NY, USA, genetic testing of the baby and her biological parents was performed.
Since KCNQ1 mutations had been shown to lead to familial atrial fibrillation (Chen et al. 2003) and short QT (Bellocq et al.2004) the group decided to analyze the DNA sequence of KCNQ1 in the baby girl.
The genetic testing confirmed the suspicion of a mutation in KCNQ1, with a G to A substitution at nucleotide 421 (g421a). This mutation results in substitution of valine by methionine at position 141 (V141M) adjacent to a previously described S140G mutation for familial AF, and different from theV307L-KCNQ1 mutation previously found in an adult patient with SQTS.
The mutation causes IKs channels to remain constitutively open leading to shortening of the action potential and tendency to atrial fibrillation.
First study looking specifically for the prevalence of a short QT interval in the general population
Gallagher MM, Magliano G, Yap YG, Padula M, Morgia V, Postorino C, Liberato FD, Leo R, Borzi M, Romeo F. Distribution and Prognostic Significance of QT Intervals in the Lowest Half Centile in 12,012 Apparently Healthy Persons. Am J Cardiol 2006;98:933-935
Mark Michael Gallagher and his colleagues at the Department of Cardiological Sciences, St. George’s Hospital Medical School, London, UK used an existing archive of medical information from apparently healthy subjects at the The Medical Forensic Institute of Rome, Italy to investigate the distribution and prognostic importance of shorter QT intervals.
Among 12,012 healthy Italian subjects (90.7 % male) 30 +/- 10 years old the cut-off QTc for the lowest half centile (60 subjects) was 361 msec and the shortest QTc 335 msec.
36 subjects with QTc ≤ 360 ms were followed for 7.9 +/- 4.5 years and none of them died suddenly during that period.
The QT intervals were measured manually and the end of the T wave was the point at which the T wave returned to the isoelectric line. QTc was calculated according to Bazett formula. Since Bazett formula overcorrects at slow heart rates the significantly slower heart seen in subjects with the shortest QTc should not be a surprise.
The absence of any QTc < 335 msec in this study confirms the impression that Short QT Syndrome is a distinct clinical entity involving QT intervals substantially shorter than those found in a normal population.
Description of a sub-group of Brugada Syndrome patients with a shorter-than-normal QT interval
Antzelevitch C, Pollevick GD, Cordeiro JM, Casis O, Sanguinetti MC, Aizawa Y, Guerchicoff A, Pfeiffer R, Oliva A, Wollnik B, Gelber P, Bonaros EP, Burashnikov E, Wu Y, Sargent JD, Schickel S, Oberheiden R, Bhatia A, Hsu L-F, Jaissaguerre M, Schrimpf R, Borggrefe M, Wolpert C. Loss-of-Function Mutations in the Cardiac Calcium Channel Underlie a New Clinical Entity Characterized by ST-Segment Elevation, Short OT Intervals, and Sudden Cardiac Death. Circulation 2007;115:442-449
By screening of 82 consecutive probands with clinically robust diagnosis of Brugade Syndrome at genetic laboratories in the US, Germany, France and Italy for ion channel gene mutations, 3 probands displaying ST-segment elevation in V1 through V3 and QTc </= 360msec were found among 7 who had mutations in genes encoding the cardiac L-type calcium channel. In addition in these 3 patients rate adaptation of QT interval was found to be reduced just like in patients with SQTS and Quinidine normalized the QT intervals and prevented stimulation-induced VT.
Genetic and heterologous expression studies revealed loss-of-function missense mutations in CACNA1C (A39V and G490R) and CACNB2 (S481L) encoding the α1 and β2b –subunits of the L-type calcium channel.
Patch-clamp experiments showed that the two mutations in CACNA1C and the one mutation in CACNB2b all cause a major loss of function in calcium channel activity.
This is the first report of loss-of-function mutations in genes encoding the cardiac L-type calcium channel to be associated with a familial sudden cardiac death syndrome in which a Brugada Syndrome phenotype is combined with shorter-than-normal QT intervals.
Mutation linking Short QT Syndrome to Sudden Infant Death Syndrome
Arnestad M et al.Prevalence of Long-QT Syndrome Gene Variants in Sudden Infant Death Syndrome Circulation 2007;115:361-367
In this study Arnestad et al. presents a patient with Sudden Infant Death Syndrome in whom a I274V-KCNQ1 mutation was found, and in the following study by Rhodes et al the mutation was found to cause a short QT interval phenotype:
Rhodes TE et al. Cardiac Potassium Channel Dysfunction in Sudden Infant Death Syndrome J Moll Cell Cardiol 2008;44(3):571-581
Electrophysiological data from patch clamp recordings (Chinese hamster ovary cells) showed that the I274V-KCNQ1 mutation in the presence of KCNE1 causes gain of function in Iks characterized by increased current density, faster activation, slower deactivation, and accumulation of instantaneous current during repeated stimulation.
To test the hypothesis that I274V may promote a short QT syndrome phenotype, computerized modeling of ventricular action potentials were performed comparing WT-Iks to heterozygous I274V-Iks. At all cycle length the AP was shorter for I274V-Iks supporting the prediction that I274V-KCNQ1 will cause a short QT phenotype, a plausible explanation for sudden death in an infant carrying this mutation.
Safety issue warning regarding drug-induced shortening of the QT interval
Holbrook M, Malik M, Shah RR, Valentin J-P. Drug induced shortening of the QT/QTc interval: An emergence safety issue warranting further modeling and evaluation in drug research and development. J Pharmacology and Toxicology Methods 2009;59:21-28
The first original article dealing with safety issues concerning drug induced shortening of the QT/QTc interval based upon presentations at a session held at the 2007 Safety Pharmacology Society meeting in Edinburgh.
It is concluded that it is currently not clear how much shortening of QT/QTc is required before it might be considered a safety issue and indeed, whether QT/QTc shortening is a suitable biomarker for cardiac arrhythmias. It is found to be clear, however, that with our current understanding, compounds which shorten QT/QTc will attract close regulatory scrutiny.
Proposed diagnostic criteria to facilitate clinical recognition of SQTS
Gollob MH, Redpath CJ, Roberts JD. The Short QT Syndrome. Proposed Diagnostic Criteria. J AM Coll Cardiol 2011;57:802-12
From 15 articles describing unique cases of SQTS the authors reviewed 61 SQTS cases as a basis for a scoring system to assist in making the diagnosis of SQTS.
Several possible limitations and errors in this scoring system proposed by Gollop MH et al. were, however, soon pointed out by Veltman C et al and Bjerregaard P in the following letters:
Veltman C, Borggrefe M. A ‘Schwartz score’ for short QT Syndrome. Nat Rev. Cardiol. 2011;8:251-252 The lack of a control group of ‘normal’ individuals with shorter than normal QTc intervals is pointed out and it is stressed that at best the scoring system can do is to estimate the probability of having the disease, not to make the diagnosis. Also the omission of using data from an electrophysiological study or exercise testing in the scoring system is emphasized.
Bjerregaard P. Proposed Diagnostic Criteria for Short QT Syndrome Are Badly Founded. J Am Coll Cardiol 2011;58:549-550. The author points out several errors in the collection of date to the article, and the limitations of using Bazett’s formula to correct the QT interval in patients with possible SQTS. It is the opinion of the author: “that the proposed diagnostic criteria for SQTS are poorly founded and should be used with great caution”.
Long-term follow-up of patients with Short QT Syndrome
Giustetto C, Schimpf R, Mazzanti A. Scrocco C, Maury P, Anttonen O, Probst V, Blanc J-J,Bragia P, Dalmasso P, Borggrefe M, Gaita F. Long-term Follow-up of Patients with Short QT Syndrome. J Am Coll Cardiol 2011;58:587-595
In this study 53 patients from the European Short QT Registry (75% males; median age: 26 years) were followed for 64 +/- 27 months.
A familial or personal history of cardiac arrest was present in 89%. Sudden death was the clinical presentation in 32%. The average QTc was 314 +/- 23 ms. A mutation of genes related to SQTS was found in 23 % of the probands: most of them had a gain of function mutation in HERG (SQTS1). Twenty-four patients received an ICD, and 12 patients received prophylactic treatment with hydroquinidine.
During follow-up, 2 already symptomatic patients received appropriate ICD shocks, and one had syncope. No arrhythmic events occurred in patients receiving hydroquinidin.
Long-term follow-up of a pediatric cohort with Short QT Syndrome
Villafañe J, Atallah J, Gollob MH, Maury P, Wolpert C, Gebauer R, Watanabe H, Horie M, Anttonen O, Kannankeril P, Faulkner B, Bleiz J, Makiyama T, Shimizu W, Hamilton RM, Young M-L. Long-Term Follow-Up of a Pediatric Cohort With Short QT Syndrome. JACC 2013;61:1183-1191
In this study 25 patients 21 years or younger from 15 different centers (84% males: median age: 15 years) were followed up for 5.9 years.
A familial or personal history of cardiac arrest was present in 84%. The mean QTc was 312 ms (range: 194 to 355 ms). A gene mutation associated with SQTS was identified in 5 (24%) of 21 probands. Ten patients received medical treatment, mainly with quinidine. Eleven of 25 index cases underwent ICD implantation.
During follow-up two patients had appropriate ICD shocks, while inappropriate shocks were observed in 64 % of patients
Genetically reduced function of an anion exchanger as a novel mechanism of SQTS
Thorsen K, Dam VS, Kjær-Sørensen K, Pedersen LN, Skeberdis A, Jurevičius J,Treinys R, Petersen IMBS, Nielsen MS, Oxvig C, Morth P, Matchkov VV, Aalkjær C, Bundgaard H, Jensen HK. Loss-of-activity-mutation in the cardiac chloride-bicarbonate exchanger AE3 causes short QT syndrome. Nat Commun 2017;8(1):1696-1706
Based upon the fact that cation channel mutations explain < 25 % of SQTS cases and other classes of genes therefore likely to be associated with the development of this disease, Thorsen K et al. in a study of two Danish families with dominantly inherited SQTS specifically looked for involvement of other genes by whole exome sequencing. In their study they identified a novel genetic etiology for SQTS in terms of a mutation in the anion exchanger Solute Carrier Family 4 Member 3 (SLC4A3) gene, which encodes a Cl,HCO3 –exchanger (AE3). The AE3 transports Cl- into the cardiomyocyte in exchange for transport of HCO3- out of the cell. The mutation leads to a trafficking defect, decreased Cl,HCO3-exchange over the cell membrane, increased pHi in combination with a decrease in Cl-I and was shown to shorten QT duration in zebrafish embryos. The authors have clearly identified a novel mutation in the cardiac chloride-bicarbonate exchanger AE3 in two independent families with SQTS, and in zebrafish demonstrated the impact of the mutation on duration of systolic contraction and on QTc duration. The authors suggest that the mechanism through which a defect in Cl,HCO3-exchange leads to shortening QTc likely relates to a combined effect of a decrease in HCO3- efflux (i.e. increased pHi) and a decrease in Cl- influx, both affecting the repolarization. It is also suggested that these findings in addition to offering insight into mechanisms of arrhythmia in general may provide new treatment possibilities in some patients with SQTS.